
Scientific journal
European Journal of Natural History
ISSN 2073-4972
ИФ РИНЦ = 0.204
GENETICS DISEASES OF THE ORGANS OF VISION IN VORONEZH REGION. LEBER OPTIC NERVE ATROPHY

Введение
Генетические патологии органов, относящихся к анализаторам, являются самыми редко встречающимися заболеваниями. В частности, самыми редкими являются болезни органов зрения. Данная статья включает клинический случай, который описывает редко встречающуюся патологию органов зрения, носящую генетическую основу. К такому заболеванию относят атрофию зрительного нерва Лебера.
Атрофия зрительного нерва Лебера – заболевание с преимущественным поражением митохондриального аппарата, которое является специфическим к клеткам сетчатки, снижая их способность вырабатывать достаточное количество энергии, необходимое для нормального функционирования нервной ткани. Чаще наследование происходит по материнской линии.
Причиной этого заболевания является мутация в митохондриальной ДНК, которая приводит к нарушению энергообеспечения нерва, из-за снижения активности ферментов митохондрий. По мимо изменений в ДНК, есть факторы, которые являются «пусковыми» в развитии заболевания: стресс, курение, употребление алкогольных напитков, черепно-мозговая травма, воздействие наркотических веществ, интерфероны, стероидные гормоны, противотуберкулезные антибиотики, любое острое соматическое заболевание и другие факторы, способствующие возникновению данной патологии [1].
В основе патогенеза лежит кодирование информации о синтезируемых белках и ферментах молекулой ДНК. В результате нарушения, произошедшего в ДНК, снижается синтез белков цепи переноса митохондрий. Из-за изменения структуры ДНК искажаются процессы транскрипции и трансляции, в результате чего синтезируются либо отличные по структуре белки и выполняющие другие функции, либо белки, необходимые для процессов образования энергии в клетках вообще не синтезируются. К таким белкам можно отнести редуктазы, убихиноны и другие белки первого комплекса митохондрий. Эти белки играют роль транспортеров молекул АТФ, а также эти соединения участвуют в метаболизме веществ с дальнейшей выработкой энергии. При снижении их выработки падает процесс синтеза АТФ в клетке. И как следствие клетки зрительных нервов испытывают гипоэрготическое состояние или состояние нехватки энергии. Следовательно, нарушается функция нерва в целом, зрение начинает падать и в конечном счете нерв перестает функционировать. Происходит его атрофия и склерозирование тех участков в головном мозге, ответственных за принятие импульса с рецепторов сетчатки [2].
Для своевременной диагностики и расширения возможности этиологической и патогенетической терапии редко встречающихся генетических аномалий органов зрения необходимо уделять особое внимание клинической картине заболеваний у каждого отдельно взятого пациента. Как правило, лечения наследственной оптической невропатии Лебера не существует, но можно замедлить прогрессирование заболевания, путем приема различных витаминов и коферментов.
В связи с этим, основной целью исследования является описание клинического случая заболевания атрофии зрительного нерва Лебера, которая из большинства заболеваний органов зрительного аппарата, является самой редкой.
Материалы и методы исследования
Исследование проводилось на базе БУЗ ВО «Воронежской областной клинической больницы №1» в отделении медико-генетической консультации перинатального центра. Основой для составления статистики по частоте встречаемости данной патологии являлись медицинские карты, которые рассматривались за последние полгода 2023 года.
Согласно полученным данным частота встречаемости больных с атрофией зрительного нерва Лебера за последние 6 месяцев на базе БУЗ ВО ВОКБ №1 составила 9,09%, т.е. 1 случай из 11 обратившихся больных с подозрением на аномалию Лебера (таблица). Наряду с этим количество больных, обратившихся в стационар больницы по поводу приобретенных патологий, таких как миопия, помутнение хрусталика, повышенное внутриглазное давление и других заболеваний, составило около 223 человек, что в 20,27 раз больше, чем количество людей, с генетическими отклонениями зрительного аппарата. Что подтверждает редкость этой патологии в клинической практике и практике врачей-генетиков.
Исследование методом мультиплексной лигазной полимеразной цепной реакции (MLPA), один из часто используемых методов для подтверждения большинства генетических патологий.
Частота встречаемости атрофии зрительного нерва Лебера в практике БУЗ ВО «Воронежской областной клинической больницы №1»
|
Приобретенные патологии органов зрения, количество больных |
Общее количество больных, с предполагаемыми генетическими аномалиями зрения |
Количество больных атрофией зрительного нерва Лебера за последние 6 месяцев 2023 года |
|
223 |
11 |
1 |
MLPA – это метод молекулярной генетики, служащий для определения относительного количества копий участков ДНК. Используется для подтверждения мутации в гене MT-ND4, которая и характеризует атрофию зрительных нервов Лебера [3]. Метод MLPA проводится следующим образом, сначала забирают кровь в пробирку с антикоагулянтом. На первом этапе самого анализа проводится разрушение ДНК и ее «слияние» со специфическими зондами. Зонды располагаются близко друг к другу. И к одному из их концов присоединена комбинация нуклеотидов или последовательность для прикрепления универсального праймера. На следующем этапе проведения анализа происходило расщепление проб с помощью фермента – лигазы. Далее производилась мультиплексная полимеразная цепная реакция-амплификация с использованием универсальных праймеров, при этом многократно увеличивается количество только лигированных фрагментов. В результате реакции амплификации получаются продукты, которые имеют разные размеры, они будут сепарироваться или отделяться с помощью капиллярного электрофореза. На компьютере будет отображаться пик и его высота, которые на электрофореграмме отражают состояние данного генетического локуса. То есть произошла ли делеция, дупликация или перед нами норма [4].
Обычно, для определения наследственной оптической невропатии Лебера, в комбинации с методом MLPA идет секвенирование по Сенгеру [5]. Для начала, у исследуемого забирают кровь в пробирку объемом около 4-5 миллилитров, содержащую этилендиаминтетрауксусную кислоту, выступающую в роли антикоагулянта. ДНК разделяют на 4 отдельные реакции секвенирования, содержащие все четыре дезоксинуклеотида (dATP, dCTP, dGTP, dTTP) и ДНК-полимеразу, достраивающую комплементарные цепи. В результате достраивания цепи можно получить на мониторе компьютера точную картину, в которой видны обрывы цепи из-за наличия мутации в ее ДНК [6].
Клинический случай
В июне 2023 года на прием в БУЗ ВО ВОКБ №1 в офтальмологическое отделение поступила женщина 30 лет с жалобами на резкое падение зрения. Из анамнеза: считает себя больной с августа 2022 года, когда на фоне полного здоровья стало постепенно снижаться зрение. В августе 2022 года стала появляться пленка перед глазами с обоих сторон. Врач-офтальмолог в августе 2022 года определил остроту зрения: справа 0,6-0,7, а слева – 0,02. Был поставлен предварительный диагноз прогрессирующая миопия. С этого момента зрение постепенно ухудшалось. В июне 2023 года офтальмолог направил женщину на обследование, для уточнения неврологического статуса пациентки. При обследовании у врача невролога было установлено: высшие мозговые центры у пациентки не нарушены, за молоточком при исследовании больная не следит (так как не видит его), объем движения глазных яблок не ограничен (смотрит по просьбе врача вверх, вниз, вправо и влево), птоза и нистагма не наблюдалось, мимика симметрична, парезов нет, сухожильные и периостальные рефлексы с конечностей равномерные живые, язык по средней линии, чувствительность (болевая и мышечно-суставное чувство) не нарушена. В пробе Ромберга устойчива, указательные пробы выполняет удовлетворительно. Пока происходили дообследования, врач-офтальмолог при повторном осмотре глазного дна через несколько дней наблюдал следующую картину: отек и бледность зрительного диска и точечные кровоизлияния в сетчатку глаза. Наследственный анамнез отягощен: мать больной в возрасте 28-29 лет потеряла зрение на оба глаза, в 32 года умерла во сне (причина неизвестна). У сына пациентки глухота с 3 лет – ему был установлен кохлеовестибулярный имплант.
В качестве инструментальных методов исследования больной было назначено МРТ головного мозга с внутривенным контрастированием. Была обнаружена картина единичного супратенториального очага глиоза (разрастания соединительной ткани в правой лобной доле, сосудистого генеза), интракраниальная асимметрия сегментов позвоночных артерий и сужение правой позвоночной артерии. Исходя из картины МРТ и неврологического статуса пациентки врач-офтальмолог назначил генетический тест ДНК методом MLPA, для подтверждения патологии атрофии зрительного нерва Лебера, с дальнейшим направлением пациентки в отделение медико-генетической консультации.
Результаты исследования и их обсуждение
По результатам метода MLPA была выявлена мутация m.11778G>A в гене MT-ND4 в гомоплазмическом состоянии. Этот ген ответственен за синтез специализированных белков первого комплекса дыхательной цепи митохондрий, к ним относятся различные виды редуктаз, убихиноны и другие белки данного комплекса. Это обуславливает нарушение процессов транспорта тверждение лабораторных и инструментальных методов, указавших на генетически обусловленную атрофию зрительного нерва Лебера, был проведен так же анализ родословной. После составления схемы родословной, которая представлена на рисунке, был проведен ее тщательный анализ врачами-генетиками.
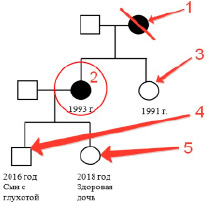
Схема родословной пациентки: 1 – мать пациентки, 2 – пациентка, 3 – сестра пациентки, 4 – сын пациентки, 5 – дочь пациентки
В родословной пациентки с ее слов, мама теряла зрение точно также как и она, следовательно она являлась носительницей мутантного гена, локализованного в митохондриальной ДНК. У пациентки имеется родная сестра, рожденная в 1991 году, у которой никаких отклонений в здоровье не наблюдалось. У больной были рождены двое детей: мальчик – родившийся в 2016 году и обладающий глухотой, и девочка, родившаяся в 2018 году, на данный момент не имеющая в здоровье никаких отклонений. Но высок риск передачи генов, ответственных за данную патологию как девочке, так и мальчику, так как передача происходит по материнской линии. Риск в потомстве высокий и стремится к 100%. Были даны следующие рекомендации: провести анализ ДНК сыну и дочери, осмотр в отделении медико-генетической консультации в динамике. Так же было назначено лечение: коэнзим Q10 (кудевита 30 mg внутрь 2 раза в день по 1 таблетке), L-карнитина постоянно в стандартных дозировках (элькар по 2,5 ml внутрь 3 раза в день).
Заключение
Таким образом, мутация, которую удалось обнаружить у женщины является, пожалуй, самой агрессивной и наиболее тяжелой формой данной патологии m.11778G> A – самая тяжелая форма атрофии зрительного нерва Лебера, а например m.3460G> A – более легкая форма. Так же существуют больные с еще одним видом мутации митохондриальной ДНК m.14484T> C – она дает наиболее благоприятный прогноз лечения.
Исследуя заболеваемость органов зрения, которые носят генетическую основу на базе БУЗ ВО «Воронежской областной клинической больницы №1», удалось выяснить, что наиболее редко встречающейся генетической патологией органов зрения является нейропатия Лебера, в данной клинике встречаемость составляет 9,09 % из обратившихся 11 человек по подозрению на эту патологию, то есть только у одного из них это заболевание подтвердилось. Так же эта патология имеет различный вид проявлений. Эта болезнь может проявляться с рождения, а может и не проявиться, и быть в форме носительства. Но при действии определенных факторов, таких как стресс, курение, алкоголь, возможно проявление атрофии зрительного нерва Лебера с быстрым прогрессирующим течением.
Данный клинический случай показывает, что осведомленность о данной болезни повышает информированность врача-клинициста в данной области, позволяет провести полное обследование, направить пациента на генетические исследования, своевременно установить диагноз, а так же назначить необходимое лечение, которое будет направлено на замедление прогрессирования заболевания.
Библиографическая ссылка
Свиридов Д.В., Ануфриева Е.И., Макеева А.В. ГЕНЕТИЧЕСКИЕ ЗАБОЛЕВАНИЯ ОРГАНОВ ЗРЕНИЯ В ВОРОНЕЖСКОЙ ОБЛАСТИ. АТРОФИЯ ЗРИТЕЛЬНОГО НЕРВА ЛЕБЕРА // European Journal of Natural History. 2024. № 5. ;URL: https://world-science.ru/en/article/view?id=34402 (дата обращения: 23.06.2026).